DOI:https://doi.org/10.65281/734537
Lazhar BOUCHLALEG 1, ZIDANI Sara 2, BAIRA Fayçal 3, Yamina Benkrima 4
1 Innovation Laboratory of Construction Eco-design and Geny Seismic (LICEGS Laboratory), Faculty of Technology, University of Batna 2, Batna 05000, Algeria
Email: [email protected] ; Orcid : 0009-0009-3680-9761
2university of Batna Departement of Food Technology, laboratory of Food Science (lSa), institute of veterinary and agricultural Sciences, 1 hadjlakhdar, alleys may 19 Biskra avenue, Batna, 05000, Algeria
Email: [email protected] ; Orcid: 0009-0004-3604-2437
3 university of Batna 2, Faculty of Technology, Department oF Sciences and Technology, alleys 53, constantine avenue. Fésdis, Batna 05078, Algeria
Email: [email protected] ; 0009-0008-0869-8614
4 MPMMDMEER Laboratory, ENS Ouargla, 30000 Ouargla, Algeria
Email: [email protected] ; Orcid: 0000-0002-7089-7904
Received: 11 November 2025 ; Accepted: 25 March 2026
Abstract
Hydantoin and thiohydantoin derivatives constitute an important family of nitrogen-based heterocyclic compounds that have attracted considerable attention because of their diverse pharmacological properties, including anticonvulsant, antimicrobial, anticancer, and enzyme inhibitory activities. In this investigation, an integrated computational strategy combining QSAR modeling, quantum chemical calculations, and molecular docking was employed to analyze the structural factors influencing their biological behavior.Eighteen hydantoin analogues reported as plasmin inhibitors were selected for the study. QSAR analysis using multiple linear regression demonstrated that molecular descriptors associated with surface area, molar refractivity, and polarizability positively affect inhibitory activity, whereas excessive molecular volume and lipophilicity contribute negatively.
Quantum chemical studies performed using HF and DFT approaches generated structural and electronic parameters that were in close agreement with experimental observations, supporting the reliability of the theoretical methodology. The presence of substituents such as methyl and chloro groups was shown to markedly influence charge distribution and molecular reactivity.Docking experiments carried out on the Metabotropic glutamate receptor 1 (GRM1, PDB ID: 5KZQ) revealed favorable ligand–receptor interactions, with binding affinities reaching approximately −9.5 kcal/mol. The stability of the complexes was mainly attributed to hydrogen-bond formation and hydrophobic interactions within the receptor active pocket.
The combined results suggest that hydantoin-based molecules represent promising scaffolds for plasmin inhibition and potential anti-epileptic applications. This work provides valuable molecular insights that may support the rational development of new therapeutic agents.
Keywords: Hydantoin derivatives; QSAR modeling; Molecular docking; Plasmin inhibition; Epilepsy; GRM1 receptor; Rational drug design.
1. INTRODUCTION
Five-membered nitrogen-containing heterocycles, particularly hydantoins and thiohydantoins, play a significant role in medicinal chemistry because of their wide range of biological and pharmacological applications. Their core structure, derived from the 2,4-imidazolidinedione nucleus, offers several positions suitable for chemical modification, allowing optimization of physicochemical and therapeutic properties.Among the best-known hydantoin-based drugs, Phenytoin remains a classical anticonvulsant widely used in the treatment of epilepsy5 ,6. Structural alterations of the hydantoin ring, especially oxygen-to-sulfur substitution yielding thiohydantoins, can substantially modify electronic distribution, lipophilicity, and biological performance.In addition to their anticonvulsant potential, hydantoin derivatives have emerged as attractive candidates for plasmin inhibition. Plasmin, a serine protease involved in fibrinolysis, tumor progression, and metastasis, represents an important therapeutic target in several pathological conditions10 ,11. Consequently, the design of effective plasmin inhibitors has become an active area of pharmaceutical research1, 2, 36.Recent studies have also emphasized the critical role of glutamatergic neurotransmission in neurological disorders. In this context, the Metabotropic glutamate receptor 1 (GRM1) has attracted considerable interest because of its involvement in synaptic regulation and neuronal excitability associated with epilepsy.The rapid evolution of computational chemistry has greatly facilitated modern drug discovery. Computational techniques such as quantitative structure–activity relationship (QSAR) analysis, density functional theory (DFT), Hartree–Fock (HF) calculations, and molecular docking provide complementary information regarding molecular geometry, electronic structure, and ligand–target interactions.Therefore, the present study employs an integrated in silico methodology combining QSAR modeling, quantum chemical investigations, and docking simulations to better understand the structure–activity relationships of hydantoin derivatives and identify promising candidates with potential plasmin inhibitory and anti-epileptic properties26,27.
2. MATERIALS AND METHODS
A dataset composed of eighteen hydantoin derivatives with experimentally determined plasmin inhibitory activities expressed as pIC₅₀ values was collected from previously published studies.
The molecular structures were initially constructed and subjected to geometry pre-optimization using molecular mechanics (MM+) methods. Semi-empirical optimization was subsequently performed with the PM3 approach to obtain energetically stable conformations. Higher-level quantum chemical calculations were then carried out using both Hartree–Fock (HF) and density functional theory (DFT) methods, particularly the B3LYP functional combined with suitable basis sets, in order to generate accurate structural and electronic descriptors12 ,13.
Several physicochemical parameters were calculated for each derivative, including molecular surface area, molecular volume, lipophilicity (log P), hydration energy, molar refractivity, and polarizability. These descriptors were selected because of their relevance to steric, hydrophobic, and electronic molecular properties.
To establish quantitative relationships between molecular structure and biological activity, multiple linear regression (MLR) analysis was applied. The statistical quality and predictive reliability of the developed QSAR models were evaluated using parameters such as the coefficient of determination (R²), Fisher statistic (F), standard error (SE), and leave-one-out (LOO) cross-validation procedures13, 18
In addition, vibrational frequency calculations were performed and compared with available experimental spectra to validate the theoretical models and confirm the accuracy of the optimized geometries.
Molecular docking studies were conducted using AutoDock Vina against the Metabotropic glutamate receptor 1 (PDB ID: 5KZQ). The resulting ligand–receptor complexes were analyzed according to hydrogen bonding, hydrophobic interactions, π–π stacking, and electrostatic contacts within the active site33, 35.
2.1 Dataset for QSAR Investigation
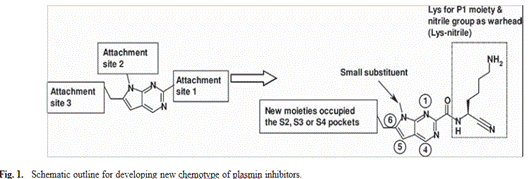
The present work focused on a structurally diverse series of hydantoin derivatives reported as plasmin inhibitors. The molecular design strategy aimed to optimize interactions within the catalytic binding pocket of the target enzyme through systematic scaffold functionalization. Different substitution positions were introduced around the heterocyclic core in order to modulate steric, hydrophobic, and electronic characteristics while maintaining favorable molecular recognition.The designed compounds were expected to interact efficiently with the S2–S4 sub-sites of plasmin through a combination of hydrogen bonding, electrostatic attractions, and hydrophobic contacts19. A lysine fragment positioned at the P1 site was incorporated to improve enzyme recognition, whereas the nitrile functionality served as an electrophilic center capable of reversible covalent interaction with the catalytic serine residue. This rational design approach combines selective pocket recognition with optimized steric complementarity to improve inhibitory potency and selectivity17, 18.
Structure–Physicochemical Property Relationships
At the beginning of the investigation, several important physicochemical descriptors were analyzed for the eighteen hydantoin derivatives in order to better understand the molecular factors influencing plasmin inhibitory activity. These descriptors were selected to represent steric, hydrophobic, and electronic molecular contributions.Molecular dimensions were characterized through molecular volume and surface area, corresponding to the spatial region enclosed by the Van der Waals surface of each compound15, 16. These parameters provide useful information regarding steric accessibility and the capacity of the ligands to fit into biological receptor cavities.Polarizability was considered an essential electronic descriptor because it reflects the ability of the electronic cloud to undergo deformation under an external electric field. Highly polarizable molecules generally exhibit stronger intermolecular interactions, particularly dispersion forces, which may enhance ligand–target affinity.Molar refractivity, which is closely associated with molecular size and electronic distribution, was also evaluated as an additional steric and electronic descriptor. Variations observed among the compounds were consistent with differences in substituent size and chemical nature. Molecules containing bulky substituents exhibited increased refractivity and polarizability values, indicating greater electronic flexibility, whereas smaller derivatives showed more compact electronic distributions.Hydration energy analysis revealed that increasing hydrophobicity leads to reduced solvation effects17,21. Since molecular recognition in biological systems largely depends on reversible interactions with surrounding water molecules, hydration behavior represents an important determinant of biological performance.The hydrogen bond donor (HBD) and hydrogen bond acceptor (HBA) capacities also varied among the investigated compounds. Molecules possessing larger numbers of donor and acceptor groups are generally capable of forming stronger and more stable intermolecular interactions with biological targets.Lipophilicity, represented by the log P parameter, was identified as a major factor influencing pharmacokinetic behavior. Moderate lipophilicity is usually associated with an appropriate compromise between membrane permeability and aqueous solubility. Excessive lipophilic character may reduce solubility, whereas highly hydrophilic molecules may exhibit insufficient membrane penetration.Overall, the obtained results indicate that biological activity strongly depends on achieving a balanced combination of molecular size, polarity, hydrogen-bonding capability, and lipophilic character within the hydantoin scaffold1,10.
Quantitative Structure–Activity Relationship (QSAR) Analysis
In the second phase of the study, quantitative structure–activity relationship analysis was employed to investigate the relationship between molecular descriptors and plasmin inhibitory activity of the hydantoin derivatives. Multiple linear regression (MLR) methodology was applied to the dataset composed of eighteen compounds using the previously calculated physicochemical parameters as independent variables14.The development of a reliable QSAR model requires both structural diversity and the selection of meaningful molecular descriptors capable of accurately representing steric, hydrophobic, and electronic effects. Since molecular descriptors are numerical representations of chemical properties, identifying the most relevant subset is essential to improve model robustness and predictive capacity.To achieve this objective, Pearson correlation analysis was performed using SPSS Statistics 19. This statistical screening procedure enabled the identification of the most significant descriptors, which were subsequently incorporated into the final MLR-QSAR model for biological activity prediction.
3. RESULTS AND DISCUSSION
3.1 QSAR Analysis of Hydantoin Derivatives as Plasmin Inhibitors
The QSAR investigation demonstrated that the inhibitory potency of hydantoin derivatives toward plasmin is strongly governed by steric, hydrophobic, and electronic molecular characteristics. Multiple linear regression analysis produced a statistically reliable predictive equation correlating the experimental pIC₅₀ values with selected molecular descriptors.The developed QSAR relationship is represented by:
pIC50=5.023+0.024S−0.025V+0.46RF+0.147Pol−0.080logP
pIC_{50}=5.023+0.024S-0.025V+0.46RF+0.147Pol-0.080\log
PpIC50 =5.023+0.024S−0.025V+0.46RF+0.147Pol−0.080logP
where:S corresponds to molecular surface area, V represents molecular volume, RF denotes molar refractivity, Pol indicates polarizability, log P represents lipophilicity.
The positive coefficients associated with surface area, refractivity, and polarizability indicate that increased steric exposure and enhanced electronic flexibility favor stronger ligand–enzyme interactions. In contrast, excessive molecular volume may negatively influence activity because of steric hindrance within the receptor active site7, 8.
Likewise, highly lipophilic derivatives may exhibit reduced biological efficiency due to unfavorable balance between solubility and target accessibility1, 3.
These observations suggest that efficient plasmin inhibitors should possess moderate molecular dimensions, sufficient polarizability, balanced hydrophilic/lipophilic properties, and appropriate surface area capable of promoting intermolecular recognition. Consequently, suitable substitution around the hydantoin nucleus can significantly modulate inhibitory activity and pharmacological performance continue4,6.
3.2 Screening of Synthesized Compounds and ADME EVALUATION
The physicochemical and pharmacokinetic properties of the synthesized hydantoin derivatives were further evaluated using the SwissADME platform. Important molecular parameters including molecular weight, oral bioavailability, skin permeability, and blood–brain barrier (BBB) permeability were analyzed in order to estimate the drug-likeness and pharmacokinetic behavior of the investigated compounds.The obtained ADME profiles indicated that most compounds possess physicochemical characteristics compatible with acceptable pharmacological performance. Moderate molecular weight and balanced lipophilicity were found to contribute positively to membrane permeability and oral bioavailability28, 42. In addition, several compounds demonstrated the potential to cross the blood–brain barrier, which is an important requirement for anti-epileptic agents targeting the central nervous system.The IUPAC nomenclature and structural characteristics of representative compounds are summarized in the corresponding tables and molecular structure figures.
Table-1: List of Compounds and their IUPAC name
| Compound number | IUPAC name |
| A | 3-[(2,4-dioxo-8-(3-chlorophenyloyl)-1,3,8- triazaspiro[4.6]undec-3- yl)methyl]benzonitrile |
| B | 3-[(2,4-dioxo-8-(3- fluorophenyloyl)-1,3,8- triazaspiro[4.6]undec-3- yl)methyl]benzonitrile |
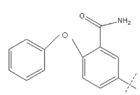
Two-dimensional Structure of the Compounds
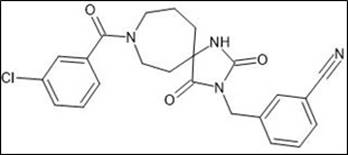
Figure 1: 3-[(2,4-dioxo-8-(3-chlorophenyloyl)-1,3,8- triazaspiro[4.6]undec-3- yl)methyl]benzonitrile
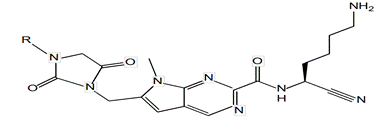
Figure 2: 3-[(2,4-dioxo-8-(3-fluorophenyloyl)-1,3,8- triazaspiro [4.6]undec-3- yl)methyl]benzonitrile
3.3 Geometry Optimization of Hydantoin and Thiohydantoin Derivatives
Geometry optimization studies were carried out using both Hartree–Fock (HF) and density functional theory (DFT) methods. The calculated structural parameters showed good agreement with available experimental measurements, confirming the reliability of the theoretical approach employed in this work.The substitution of oxygen atoms by sulfur within the heterocyclic ring significantly influenced molecular geometry and electronic properties. In particular, sulfur-containing derivatives exhibited longer bond lengths, increased molecular softness, and higher polarizability values. These modifications are expected to improve intermolecular interactions with biological targets by enhancing electronic flexibility.Frontier molecular orbital analysis demonstrated that molecular reactivity is strongly related to the HOMO–LUMO energy gap. Molecules possessing smaller energy gaps generally showed higher chemical reactivity and greater interaction potential. Electron-withdrawing substituents tended to favor electrophilic behavior, whereas electron-donating groups promoted nucleophilic character29, 42.Vibrational frequency calculations confirmed the presence of characteristic absorption bands associated with N–H, C=O, and C=S functional groups. The theoretical spectra were consistent with experimental observations, supporting the validity of the optimized molecular structures.Overall, the combined quantum chemical and physicochemical analyses revealed that parameters such as molecular size, hydrogen-bonding capability, polarizability, and lipophilicity play essential roles in determining plasmin inhibitory activity and receptor binding efficiency. Docking investigations additionally confirmed favorable interactions with the GRM1 receptor, highlighting the potential anti-epileptic properties of the studied compounds.The three-dimensional conformations and atom numbering systems adopted for hydantoin derivatives were generated using GaussView 091,2,3.
Conformation 3D of molecular structure and atom numbering adopted in this study for hydantoin (GaussView 09)
Experimental and calculated values of bond lengths and bond angles of Hydantoin
Experimental and calculated vibrational spectra of hydantoin
Table I. Chemical structures, physicochemical properties and experimental activities of Hydantoin derivatives.
| Comp. | R | Plasmin pIC50exp. [27] | pIC50 pred. | pIC50 resd. | SAG Å2 | MV Å3 | MW (a.m.u) | Log P | HE (Kcal/mol) | RF Å3 | Pol Å3 | |
| 1 | 3.880 | 3.825 | 0.060 | 816.39 | 1380.00 | 492.51 | −2.11 | −15.16 | 134.36 | 49.57 |
2 3.745 3.650 0.094 840.86 1438.22 504.44 −2.50 −17.18 140.61 52.13
3 3.481 3.356 0.125 890.12 1522.15 516.60 −0.56 −13.40 147.72 55.16
4 3.796 3.907 -0.111 860.97 145,40 517.55 -2.99 -20.00 142.05 52
5 3.678 3.801 −0.123 845.88 1428.68 499.53 −1.78 −19.61 139.21 51.51
6 3.602 3.692 −0.089 905.55 1519.71 532.56 −2.66 −17.95 145.35 54.05
7 3.585 3.600 −0.015 859.82 1455.11 539.57 −3.61 −24.56 142.59 50.31
8 3.603 3.509 0.093 881.13 1498.23 553.60 −3.36 −22.19 147.49 52.15
9 3.658 3.761 −0.104 810.85 1377.26 485.51 −1.69 −19.23 134.83 49.67
10 4.000 3.928 0.072 847.36 1431.54 502.53 −3.09 −18.35 139.52 51.81
11
3.398 3.542 −0.144 904.35 1522.69 535.56 −3.65 −23.22 143.58 53.76
12
3.886 3.784 0.101 871.06 1463.28 515.53 −2.62 −23.91 140.92 52.15
13
4.292 4.302 −0.009 921.33 1560.45 552.59 −2.26 −20.53 160.30 58.
14 4.194 4.066 0.127 996.76 1689.94 596.65 −2.85 −22.08 171.27 62.43
15 4.060 3.9363 0.124 950.20 1613.59 567.61 −3.21 −21.83 160.89 59.24
16 4.004 4.0632 −0.058 964.04 1649.34 595.62 −3.74 −24.98 168.12 61.39
17 3.292 3.6261 −0.333 994.68 1714.24 609.64 −3.34 −24.01 172.72 63.23
18
3.770 3.5765 0.193 991.00 1705.22 612.65 −4.72 −25.04 167.90 62.71

Correlation Between Biological Activity and Molecular Descriptors
The statistical relationship established between the experimental biological activities and the selected molecular descriptors can be expressed by the following equation:
pIC50=5.023+0.024S−0.025V+0.46RF+0.147Pol−0.080logP
pIC_{50}=5.023+0.024S-0.025V+0.46RF+0.147Pol-0.080logP
pIC50=5.023+0.024S−0.025V+0.46RF+0.147Pol−0.080logP
The statistical parameters obtained for the model were:Number of compounds (n) = 18
Correlation coefficient (r) = 0.868 ,Fisher statistic (F) = 7.351 ,Standard error (SE) = 0.159
These values demonstrate that the developed QSAR model provides a satisfactory correlation between structural descriptors and plasmin inhibitory activity. The high correlation coefficient indicates strong agreement between experimental and predicted values, while the Fisher statistic confirms the statistical significance and predictive reliability of the model.
The negative coefficients associated with molecular volume and lipophilicity indicate that excessive steric bulk and hydrophobicity reduce biological activity. Conversely, larger surface area, increased refractivity, and enhanced polarizability contribute positively to inhibitory potency by improving intermolecular recognition and electronic interactions.Validation of the model through leave-one-out (LOO) cross-validation confirmed its predictive stability. The PRESS value was lower than the sum of squared deviations (SSY), demonstrating that the model performs significantly better than random prediction. Moreover, the correlation between predicted and experimental activities remained satisfactory, with acceptable R² and r² values.Residual analysis did not reveal any systematic deviation, supporting the robustness and consistency of the developed QSAR equation. The predicted pIC₅₀ values were found to be in good agreement with experimental measurements, further confirming the predictive capability of the model.
Fig. 2. Predicted plots versus experimental observed specific plasmin activity of hydantoins
The QSAR model showed statistical significance at the 95% confidence level, indicating reliability. The negative coefficients of molecular volume (V) and lipophilicity (log P) suggest that increasing these descriptors decreases biological activity, while higher surface area, refractivity, and polarizability enhance plasmin inhibitory activity. Model validation using leave-one-out (LOO) cross-validation confirmed its predictive ability, with PRESS lower than SSY, indicating better-than-chance performance. The model exhibited acceptable correlation with experimental data (R² = 0.754; r² = 0.651). Residual analysis showed no significant systematic error, supporting model stability. Predicted pIC₅₀ values obtained from the QSAR equation agreed well with experimental values, and the regression plot confirmed satisfactory predictive performance of the developed model.
3.6 Molecular Docking Study Against the GRM1 Receptor
3.6 Molecular Docking Study Against the GRM1 Receptor
Molecular docking simulations were performed to evaluate the binding interactions between the synthesized hydantoin derivatives and the Metabotropic glutamate receptor 1 (GRM1) using the crystallographic structure identified by PDB ID: 5KZQ. The obtained docking results demonstrated favorable ligand–receptor affinity, with calculated binding energies approaching −9.5 kcal·mol⁻¹, indicating the formation of stable molecular complexes.The stability of the docked conformations was mainly attributed to hydrogen-bond interactions involving amino acid residues such as SER128, ARG40, THR151, THR199, and SER255. Additional stabilization originated from hydrophobic contacts and π–π stacking interactions established within the receptor active cavity.The introduction of aromatic substituents was found to improve binding affinity by increasing π-surface interactions and enhancing steric complementarity between the ligand and receptor binding pocket. These observations suggest that hydantoin derivatives may act as promising modulators of glutamatergic signaling pathways associated with epileptic disorders.
3.7 Comparative Analysis of Hydantoin and Thiohydantoin Analogues
A comparative evaluation of docking behavior and intermolecular interactions revealed distinct binding characteristics between hydantoin and thiohydantoin analogues.Thiohydantoin derivatives displayed enhanced hydrophobic interactions due to the replacement of oxygen by sulfur atoms, which increases lipophilic character and electronic softness. In contrast, classical hydantoin compounds retained stronger hydrogen-bonding capability because of the presence of carbonyl oxygen atoms capable of acting as efficient hydrogen-bond acceptors.These complementary interaction profiles suggest two different optimization strategies for future molecular design. Hydantoins appear more suitable for receptor environments dominated by polar interactions and hydrogen bonding, whereas thiohydantoins may exhibit improved performance in hydrophobic binding pockets1,3,4.
3.8 Implications for Drug Design
The combined results obtained from QSAR modeling, quantum chemical calculations, and molecular docking provide valuable information for the rational development of novel therapeutic agents based on the hydantoin scaffold.The study indicates that efficient drug candidates should combine several important molecular features, including moderate molecular size, balanced lipophilicity, sufficient polarizability, and preserved hydrogen-bond donor/acceptor functionalities. The incorporation of aromatic substituents capable of promoting π-interactions also appears beneficial for improving receptor affinity and molecular recognition.These structural characteristics collectively contribute to stronger ligand–target interactions, enhanced binding stability, and improved steric complementarity within the active site of the receptor. Therefore, strategic structural modification of hydantoin derivatives may lead to compounds with optimized pharmacological profiles and increased biological activity4, 5.
Molecular Docking and Structural Validation
Docking investigations were conducted to analyze the interactions between the synthesized compounds and selected biological targets. Ligand structures were initially constructed using ChemSketch and subsequently converted into three-dimensional PDB formats suitable for docking calculations.The crystal structure of the GRM1 receptor (PDB ID: 5KZQ) was retrieved from the Protein Data Bank for computational analysis. Structural validation of the receptor was performed using Ramachandran plot analysis, which confirmed the good stereochemical quality of the protein model, with approximately 91% of amino acid residues located within favored regions.Docking calculations revealed strong ligand–protein interactions for all investigated complexes, with binding energies generally lower than −9.0 kcal/mol, indicating favorable binding stability25,27. The binding modes observed for representative compounds demonstrated the presence of multiple stabilizing interactions inside the receptor active site, including hydrogen bonds, hydrophobic interactions, and aromatic contacts.GRM1 belongs to the family of metabotropic glutamate receptors (mGluRs), which are members of group C G protein-coupled receptors (GPCRs). These receptors are widely distributed throughout the central nervous system, particularly within the cerebral cortex, cerebellum, and spinal cord, where they regulate glutamate-mediated neurotransmission.Previous pharmacological and genetic studies have demonstrated that several mGluR subtypes, especially groups II and III, exhibit anticonvulsant properties. In the present investigation, the strong binding affinity observed between the synthesized hydantoin derivatives and GRM1 suggests that these compounds may function as potential modulators ofglutamatergic signaling pathways involved in epilepsy. Consequently, the investigated molecules may represent promising candidates for the development of future anti-epileptic therapies29.
Figure 3: Crystal structure of Metabotropic Glutamate receptor proteins (PDB ID: 5KZQ)
Table 2. Binding Affinity of the Docking Compounds with the selected G protein-coupled glutamate receptor signalling protein.
Figure 4: Ramachandran plot provides an overview of allowed and disallowed regions of torsion angle values, serving as an important indicator of the quality of protein three-dimensional structures
Figure5:Molecular docking interaction receptor protein (PDB ID: 5KZQ) with synthesized compound A
Figure6:Molecular docking interaction of receptor protein (PDB ID: 5KZQ) with synthesized compound B
Molecular docking studies were performed to evaluate the interactions between the synthesized compounds and target proteins, with GRM1 showing the highest degree of binding affinity. GRM1 is a member of the metabotropic glutamate receptors (mGluRs), belonging to group C of G protein-coupled receptors (GPCRs). These receptors are widely distributed in the central nervous system, including the cerebral cortex, cerebellum, and spinal cord, where they mediate glutamate-dependent signaling.Pharmacological and genetic studies have shown that mGluRs, particularly groups II and III, exhibit anticonvulsant effects. In the present study, docking results revealed strong binding of the synthesized compounds to GRM1, with average binding energies of approximately −9.5 kcal/mol for both compounds 1 and 2. These findings suggest that the designed molecules may act as potential modulators of GRM1, supporting their relevance as promising candidates for epilepsy treatment.