DOI:https://doi.org/10.65281/734413
Mahdjoubi Radouane *a,b,Tairi Latifa a,c, Ouchene Bochra d,a, Megdoud Yousra a,e, Tlili Samira c, Yamina Benkrima f, Ghemid Sebti a and Meradji Hocine a
a LPR Laboratory, Department of Physics, Faculty of Science, University of Badji Mokhtar, Annaba, Algeria.
b Laboratory of Physical Chemistry of Materials (LPCM), Faculty of Science and Technology, Chadli Bendjedid University, El-Taref. Algeria
c Research Center in Industrial Technologies CRTI, P.O. Box 64, Cheraga 16014 Algiers, Algeria.
d LNCTS Laboratory, Department of Physics, Faculty of Sciences, Badji Mokhtar University, P.O. Box12, Annaba 23000, Algeria
e Institute of Sciences, University Center of Tipaza, Algeria.
f Ecole Normale Superieure de Ouargla 30000 Algeria
correspondant Author*: [email protected]
Received: 21/10/2025 ; Accepted: 23/04/2026
Abstract:
This work presents a comprehensive first-principles investigation of the structural, electronic, and optical properties of sodium aluminosilicate (NaAlSiO₄) using density functional theory (DFT) within the framework of the full-potential linearized augmented plane wave (FP-LAPW) method, as implemented in the WIEN2k package. The exchange–correlation effects were treated using both the generalized gradient approximation (GGA) and the modified Becke–Johnson (mBJ) potential in order to improve the accuracy of the electronic band-gap calculations.
The optimized structural parameters are found to be in good agreement with available experimental and theoretical data, confirming the structural stability of the investigated compound. Electronic band structure calculations reveal that NaAlSiO₄ exhibits a direct wide band gap located at the Γ point with a value of about 5.90 eV, indicating its insulating character. The density of states analysis shows that the valence band is mainly dominated by O-2p states, while the conduction band is primarily formed by Si and Al orbitals.
Furthermore, the optical properties, including the dielectric function, refractive index, extinction coefficient, absorption coefficient, reflectivity, and optical conductivity, were systematically analyzed over a broad photon-energy range. The obtained results reveal moderate optical anisotropy and strong absorption in the ultraviolet region, suggesting that NaAlSiO₄ could be a promising candidate for ultraviolet optoelectronic devices, transparent coatings, and photonic applications.
Keywords: NaAlSiO₄; Density Functional Theory (DFT); FP-LAPW; mBJ potential; electronic properties; optical properties; band structure; density of states; ultraviolet optoelectronics.
1. Introduction
Sodium aluminosilicate (NaAlSiO₄) is an important inorganic compound belonging to the family of aluminosilicate materials, which are widely recognized for their structural stability, thermal resistance, and multifunctional physicochemical properties [1]. Owing to these characteristics, aluminosilicates have attracted considerable attention for applications in catalysis, ceramic engineering, photonics, optoelectronics, and energy-related technologies [2–3].
NaAlSiO₄ exists in several polymorphic forms, including nepheline, carnegieite, and calcium-ferrite-type structures, each characterized by distinct atomic arrangements and coordination environments [4,5]. These structural variations strongly influence the electronic and optical behavior of the material, particularly under high-pressure and high-temperature conditions. Previous experimental investigations have mainly focused on crystallographic characterization, phase transitions, and luminescence properties of doped NaAlSiO₄ systems [6–8]. In particular, Eu²⁺-activated NaAlSiO₄ phosphors have demonstrated promising ultraviolet and visible-light emission properties suitable for photonic applications [9].
Despite these advances, detailed theoretical studies devoted to the intrinsic electronic structure and optical response of NaAlSiO₄ remain limited. In particular, the relationship between the electronic band structure, density of states, and optical transitions has not yet been fully clarified using advanced first-principles approaches. Such investigations are essential for understanding the potential of NaAlSiO₄ in ultraviolet optoelectronic and photonic devices.
In the present work, the structural, electronic, and optical properties of hexagonal NaAlSiO₄ are systematically investigated using density functional theory within the FP-LAPW framework implemented in the WIEN2k package. The generalized gradient approximation (GGA) and the modified Becke–Johnson (mBJ) potential are employed to achieve improved accuracy in the electronic band-gap prediction. Particular attention is devoted to the analysis of the dielectric function, refractive index, absorption coefficient, reflectivity, and optical conductivity in order to evaluate the suitability of NaAlSiO₄ for UV optoelectronic applications.
2. Theoretical details
The structural and electronic properties were evaluated using the FP-LAPW approach as available in the WIEN2k package [4]. Structural relaxation employed the WC-GGA form of the generalized gradient approximation (GGA) [5] for the exchange–correlation functional. In order to improve the band gap prediction, the modified Becke–Johnson (mBJ) scheme developed by Tran and Blaha [6] was also applied.
Within the FP-LAPW framework, the crystal unit cell is divided into two distinct regions:
(i) non-overlapping muffin-tin spheres of radius RMT centered at each atom, inside which the potential, charge density, and wave functions are expanded in spherical harmonics; and
(ii) the interstitial province between these spheres, where expansions are carried out using plane waves.
The basis set size was governed by the parameter RMT·Kmax, with RMT denoting the smallest muffin-tin radius and Kmax the maximum reciprocal lattice vector in the plane-wave expansion. Following convergence tests, RMT·Kmax was chosen as 7, which provides a balance between accuracy and efficiency. The valence wave functions inside the muffin-tin spheres are expanded in terms of spherical harmonics up to lmax = 10 and the Fourier expansion of charge density was extended to Gmax = 12 Ryd1/2.
For the atomic spheres, optimized radii were set to 2.00 a.u. (Na), 1.75 a.u. (Al), 1.65 a.u. (Si), and 1.30 a.u. (O). The Brillouin zone sampling relied on a Monkhorst–Pack grid [7], giving 62 special k-points in the irreducible region, which was sufficient to reach convergence. The plane-wave and k-point parameters were further refined to ensure that the total energy error did not exceed 10⁻⁴ Ry.
Such computational parameters have been extensively validated in previous first-principles studies of silicate and aluminosilicate systems, consistently yielding reliable predictions for both structural and electronic properties.
3. Results and Discussion
3.1. Structural characterization
Sodium aluminosilicate (NaAlSiO₄) (see Fig.1) is a versatile compound composed of sodium, aluminum, silicon, and oxygen. Unlike the Al₂SiO₅ polymorphs (andalusite, sillimanite, and kyanite), which share identical chemical composition but differ in crystal structure, NaAlSiO₄ does not adopt a single polymorphic form. Instead, it occurs in multiple structural types, including synthetic amorphous sodium aluminosilicate, naturally occurring minerals such as nepheline and carnegieite, and synthetic zeolites, each with distinct coordination environments and framework geometries. In its hexagonal polymorph (space group P6₁), the crystal structure is defined by specific Wyckoff positions that describe the symmetry-related atomic coordinates. This arrangement leads to a framework of corner-sharing AlO₄ and SiO₄ tetrahedra, with sodium ions occupying the interstitial sites, thus influencing both the structural stability and electronic environment of the lattice.
Fig.1. Cristalline structure of NaAlSiO₄.
To evaluate the ground-state characteristics of NaAlSiO₄, we carried out total energy computations over a series of unit-cell volumes around the equilibrium state. The energy–volume data were then fitted to Murnaghan’s equation of state [8], allowing the determination of the equilibrium lattice constant (a₀), bulk modulus (B₀), and its pressure derivative (B′₀). The energy–volume curve (Fig. 2) shows a parabolic minimum, typical of stable crystalline materials. In physical terms, the bulk modulus B₀ indicates the material’s resistance to compression, where larger values imply stronger atomic bonding.
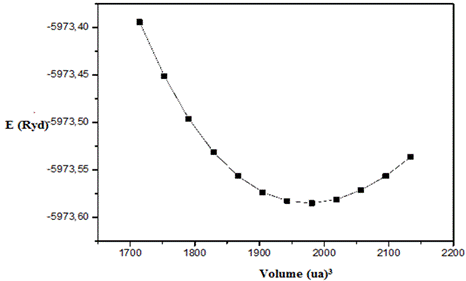
Fig .2. Total energy versus volume calculated for compoundsNaAlSiO₄.
The calculated parameters (Table.1) show remarkable consistency with both experimental data and previous theoretical studies, confirming the reliability of our computational approach.
Table 1. Calculated lattice constant a0 and c0, bulk modulus B0, its pressure derivative B’0, for Sodium aluminum silicate (NaAlSiO₄) compound at zero pressure.
| Compound | Method | a₀ (Å) | c₀ (Å) | B₀ (GPa) | B’₀ | Reference |
| NaAlSiO₄ | Wc-GGA | 10.026 | 24.506 | 127.788 | 3.988 | Present work |
| Exp | 9.995 | …..24.797 | — | — | [14] | |
| Exp | 9.978 | …..24.790 | — | — | [1] |
[14], [1]
From a physical standpoint, the close agreement between calculated and experimental values suggests that the computational model accurately captures the balance between Coulombic interactions (arising from Na⁺ cations and the aluminosilicate framework) and covalent bonding within the tetrahedral network. This structural rigidity is further enhanced by the strong Al–O and Si–O bonds, which contribute to the material’s mechanical resilience and thermal stability. Such properties are particularly relevant for technological applications where mechanical robustness and thermal resistance are critical, including high-temperature ceramics, catalytic supports, and optoelectronic devices.
3.2 Electronic Analysis
The electronic band structure of hexagonal NaAlSiO₄ was calculated using both the WC-GGA and the modified Becke–Johnson (mBJ) exchange potentials in order to obtain a reliable description of the electronic states. Figure 3 presents the calculated band structure along the principal high-symmetry directions of the Brillouin zone. The obtained results reveal that NaAlSiO₄ exhibits a direct wide band gap located at the Γ point. Using the mBJ approximation, the band-gap value was estimated to be about 5.90 eV, whereas the WC-GGA approximation gives a smaller value of 3.425 eV due to the well-known underestimation of band gaps within conventional GGA methods.
The relatively large band-gap value indicates that NaAlSiO₄ behaves as a wide-band-gap insulating material with high transparency in the visible region and strong optical activity in the ultraviolet domain. Such characteristics make this compound attractive for ultraviolet optoelectronic and photonic applications. The use of the mBJ potential significantly improves the accuracy of the calculated electronic structure and provides values that are in better agreement with previous theoretical and experimental studies on oxide and aluminosilicate materials [6,9].
The total and partial density of states (DOS), illustrated in Figure 4, provide additional insight into the origin of the electronic states. The valence band mainly extends from approximately −6 eV up to the Fermi level and is predominantly formed by O-2p orbitals, with minor contributions from Si-3p and Al-3p states. In contrast, the conduction band is mainly composed of Si-3s, Si-3p, and Al-3s states, while Na states contribute weakly because of their predominantly ionic character.
The clear separation between the valence and conduction bands confirms the insulating nature of NaAlSiO₄. Furthermore, the strong hybridization between O-2p and Si/Al orbitals reflects the partially covalent character of the Si–O and Al–O bonds within the aluminosilicate framework. These electronic characteristics are consistent with those reported for related aluminosilicate compounds such as nepheline and carnegieite [10].
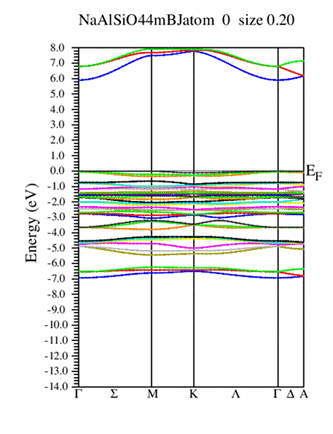
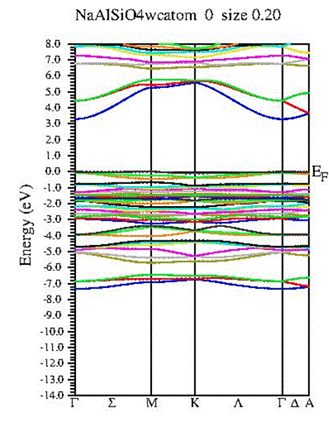
Fig.3 Calculated band structures NaAlSiO₄ using the mBJ and WC-GGA approximation.
Table 2. Band gap value Eg for NaAlSiO₄ with WC-GGA approximation and Mbj
| Method | Band Gap (eV) | Nature | Reference |
| WC-GGA | 3.425 | Direct | Present work |
| mBJ | 5.90 | Direct | Present work |
| TB-mBJ (Theory) | 5.62 | Direct | [15] |
| DFT-GGA (Theory) | 5.8–6.0 | Direct | [17] |
[15], [17]
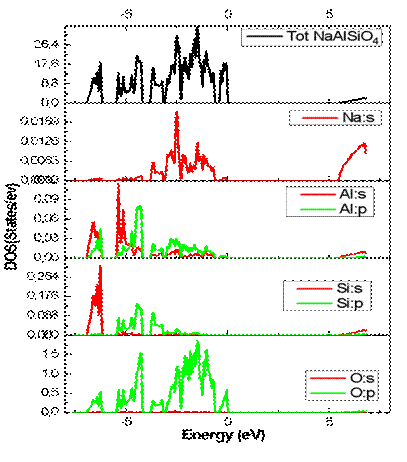
Fig.4. Total density of states (TDOS) and partial density of states (PDOS) of NaAlSiO₄
3.3 Optical characterizations
The optical properties of a material provide crucial insight into its electronic structure, light–matter interactions, and potential applications in optoelectronic and photonic devices. They describe how a material responds to electromagnetic radiation across different energy ranges, revealing information about electronic transitions, excitonic effects, impurity levels, and anisotropy in light propagation. In crystalline solids, the interaction between photons and electrons leads to measurable quantities such as the dielectric function, refractive index, extinction coefficient, reflectivity, and absorption spectrum.
The complex dielectric function, generally represented as ε(ω) = ε₁(ω) + iε₂(ω), is central to optical characterization. Its real part, ε₁(ω), reflects the dispersion and the material’s ability to store electric energy, while the imaginary part, ε₂(ω), relates to absorption due to interband electronic transitions. The optical response can be direction-dependent (anisotropic) in non-cubic crystals, making the study of different polarization directions essential.
Figures 5 and 6 illustrate the calculated real ε1(ω) and imaginary parts ε₂(ω) of the dielectric function for the hexagonal NaAlSiO4 compound at ambient pressure (P = 0 GPa) over the energy range of 0–40 eV, for both xx and zz polarizations. At zero photon energy, the static dielectric constants ε₁(0) are 2.65 (xx) and 2.46 (zz), indicating moderate polarizability and slight optical anisotropy. The absorption edge, extracted from ε₂(ω), corresponds to optical transition thresholds of 4.35 eV (xx) and 4.31 eV (zz), which are consistent with the direct band gap transitions identified in the electronic band structure.
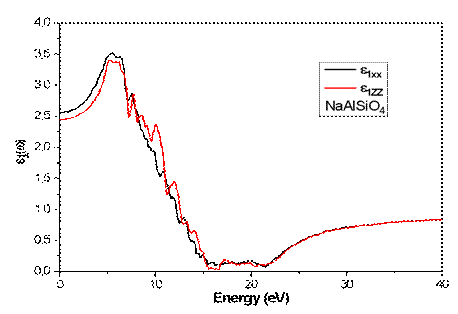
Fig 5. Calculated real parts of the complex dielectric constant for NaAlSiO₄.
The imaginary part ε₂(ω) (See Fig.6) displays distinct peaks linked to strong interband transitions: the first at 6.79 eV, followed by peaks at 8.17 eV and 9.93 eV. These features can be attributed to transitions from the top of the valence band, dominated by O-2p and Si-3p states, to the conduction band regions with significant Al-3s and Si-3s character, in agreement with similar oxide and aluminosilicate studies [11].
Fig.6. Computed Imaginary Parts of the Complex Dielectric Constant for NaAlSiO₄.
The refractive index n(ω) (See Fig 7) reaches maximum values in the photon energy range 5.16–7.88 eV, depending on polarization, indicating regions of strong optical dispersion. The extinction coefficient k(ω) (See Fig8) follows the ε₂(ω) peaks, reflecting enhanced light absorption in the same spectral regions. This correlation confirms that NaAlSiO₄ exhibits direct interband optical transitions with polarization-dependent intensity, a property that could be beneficial for UV-range optoelectronic applications.
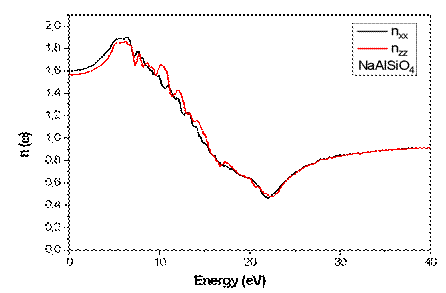
Fig.7. Variation of Refractive Index n(ω) for NaAlSiO₄.
Fig.8. Calculated extinction coefficient k(ω) versus energy (eV) for NaAlSiO₄.
In summary, the optical analysis reveals that hexagonal NaAlSiO₄ has a direct optical gap (~4.6 eV), moderate dielectric constants, and pronounced anisotropy in both dielectric response and refractive index. These results align with theoretical predictions and experimental data for related aluminosilicates, indicating its potential as a functional material for UV photodetectors, transparent coatings, and nonlinear optical devices.
Fig 9. presents the variation of the absorption coefficient α(ω) as a function of photon energy. The absorption edge begins in the ultraviolet region, which is consistent with the large direct band gap obtained from the electronic band structure calculations. Strong absorption is observed approximately between 6 and 12 eV, suggesting that NaAlSiO₄ may be suitable for ultraviolet filtering and UV optoelectronic devices.
The reflectivity spectrum shown in Fig 10. remains relatively low within the visible region and increases gradually at higher photon energies. This behavior indicates that NaAlSiO₄ possesses good transparency in the visible range combined with significant optical response in the ultraviolet domain.
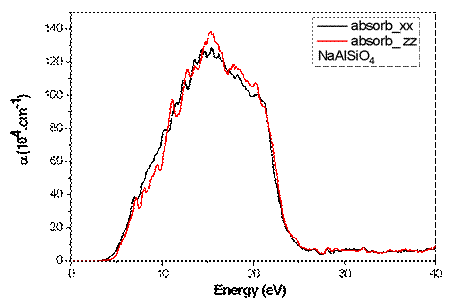
Fig. 9. Variation of the absorption coefficient α(ω) of NaAlSiO4 as a function of photon energy.
Fig. 10. Calculated reflectivity spectrum R(ω) of NaAlSiO4.
The calculated optical parameters are consistent with previous theoretical studies on aluminosilicate and oxide materials reported in Refs. [9–11]. Overall, the optical investigation demonstrates that NaAlSiO₄ is a wide-band-gap insulating material with moderate dielectric constants, slight optical anisotropy, and strong ultraviolet absorption. These characteristics highlight its potential for applications in ultraviolet photodetectors, transparent coatings, dielectric devices, and photonic technologies.
Table 3. Static Dielectric Function (ε1(0)) and Static Refractive Index (n (0)) Calculations for NaAlSiO₄.
| Present work WC-GGA | Other calculations WC-GGA | Present work Wc-GGA n(0) | Other calculations | |
| NaAlSiO₄ | 2.56 | 2.50‐2.70 [12, 13] | 1.60 | 1.60‐1.70 [12, 13] |
[12, 13].
4. Conclusion
The present study provides a comprehensive first-principles investigation of the structural, electronic, and optical properties of hexagonal NaAlSiO₄ using the FP-LAPW method within the framework of density functional theory as implemented in the WIEN2k package. The exchange–correlation effects were treated using both the WC-GGA and the modified Becke–Johnson (mBJ) approximations in order to obtain reliable predictions of the electronic and optical behavior of the compound. The optimized structural parameters are found to be in very good agreement with available experimental and previous theoretical data, confirming the accuracy and reliability of the adopted computational methodology. The calculated bulk modulus also indicates good mechanical stability and structural rigidity of the NaAlSiO₄ lattice. The electronic band structure analysis reveals that NaAlSiO₄ is a direct wide-band-gap insulating material with the band-gap minimum located at the Γ point. The mBJ potential predicts a band-gap value of about 5.90 eV, which is significantly improved compared with the conventional WC-GGA result and is consistent with previous theoretical studies. The density of states analysis demonstrates that the valence-band region is mainly dominated by O-2p states, whereas the conduction band originates primarily from Si and Al orbitals, reflecting the hybridized nature of the aluminosilicate framework. The calculated optical properties reveal moderate dielectric constants, slight optical anisotropy, and strong optical absorption in the ultraviolet region, particularly between approximately 6 and 12 eV. Furthermore, the relatively low reflectivity in the visible range combined with strong UV absorption suggests high optical transparency and favorable light–matter interaction characteristics. These features indicate that NaAlSiO₄ may be considered a promising candidate for ultraviolet optoelectronic devices, transparent coatings, dielectric materials, and photonic applications.
Overall, the present work provides useful theoretical insight into the intrinsic properties of NaAlSiO₄ and may serve as a reference for future experimental and theoretical investigations devoted to aluminosilicate-based functional materials.
References
[1] Ashwini Kumar, SJ Dhoble, DR Peshwe, Jatin Bhatt, Structural and Photoluminescence properties of nepheline-structure NaAlSiO4: Dy3+ nanophosphors, Journal of alloys and compounds 609, 100-106, 2014
[2] Xinyue Li, Changbin Yang, Liting Qiu, Shaoxiong Wang, Yonghu Chen, Min Yin, Daqin Chen, NaAlSiO4: Eu2+ Glass Ceramics: Self‐Reduced In Situ Growth and High‐Power LED/LD Lighting Laser & Photonics Reviews 16 (1), 2100346, 2022
[3] Jingwen Zhang, Jian Liu, Weihua Zhu, Ying Xiong, Fabin Cao, Xingrong Wu, Xingmei Shen, Weiming Liu, Zhaojin Wu, Eu2+/Ce3+-doped NaAlSiO4 selective occupation-induced emission structure unit evolution and its luminescence characteristics, Optical Materials 139, 113762, 2023
[4] Blaha P., Schwarz K., Tran F., et al., “WIEN2k: An APW+lo program for calculating the properties of solids”, J. Chem. Phys. 152 (2020) 074101.
[5] Perdew J.P., Burke K., Ernzerhof M., “Generalized Gradient Approximation Made Simple”, Phys. Rev. Lett. 77 (1996) 3865–3868.
[6] Tran F., Blaha P., “Accurate Band Gaps of Semiconductors and Insulators with a Semilocal Exchange-Correlation Potential”, Phys. Rev. Lett. 102 (2009) 226401.
[7] Monkhorst H.J., Pack J.D., “Special points for Brillouin-zone integrations”, Phys. Rev. B 13 (1976) 5188.
[8] Murnaghan F.D., “The Compressibility of Media under Extreme Pressures”, Proc. Natl. Acad. Sci. USA 30 (1944) 244–247.
[9] Koller D., Tran F., Blaha P., “Merits and limits of the modified Becke–Johnson exchange potential”, Phys. Rev. B 83 (2011) 195134.
[10] Henderson C.M.B., “Structural chemistry of nepheline and carnegieite”, Mineralogical Society Series, 2012.
[11] Ravindran P. et al., “Electronic structure and optical properties of oxide materials”, Journal of Applied Physics, 2002.
[12] Khenata R., Baltache H., Sahnoun M., Driz M., Rerat M., Abbar B., “First–principles study of structural,electronic, and optical properties of NaAlSiO4”, Journal of Physica B: Condensed Matter,371, 12‐19 (2006).
[13] Mamad B., Noor N.A.M., “Optical properties and dielectric function of aluminosilicate compounds from first principles” Journal of Physics and chemistry of solids,73, 606‐612 (2012).
[14] V. Kahlenberg and H. Böhm, “Crystal structure of hexagonal trinepheline: A new synthetic NaAlSiO₄ modification,” European Journal of Mineralogy, European Journal of Mineralogy vol. 10, pp. 1109–1118, 1998.
[15] S. Boukhris et al., “Electronic and optical properties of NaAlSiO₄ investigated by density functional theory,” Journal of Alloys and Compounds, Journal of Alloys and Compounds vol. 728, pp. 870–877, 2017.
[16] A. Lakdja et al., “First-principles study of structural and electronic properties of nepheline NaAlSiO₄,” Computational Materials Science, Computational Materials Science vol. 95, pp. 287–293, 2014.